Immunopepper modes
Immunopepper provides four different command-line basic modes:
Build mode: Core part of ImmunoPepper. Traverses the input splice graph and generates all possible peptides/kmers.
Samplespecif mode: This mode filters from the foreground kmer list the peptides that are also appearing in the background kmer list, and keeps only the novel kmers. It needs to be applied after the build mode.
Cancerspecif mode: This mode performs a set of filtering steps to remove kmers that are not specific to the different cancer samples. Go to the mode description for more information.
Mhcbind mode: This mode allows the user to perform MHC binding predictions on the kmers generated by Immunopepper. It is a wrapper tool for the library MHCtools, which integrates different MHC binding affinity prediction tools. It currently contains NetMHC3, NetMHC4, NetMHCpan, NetMHCIIpan, NetMHCcons and MHCflurry.
Pepquery mode: This mode allows the user to perform peptide validation on the peptides generated by cancerspecif mode. It is a wrapper tool for the software PepQuery.
The following flowchart be helpful to decide which is the best way to use immunopepper and its different modes, according to the user’s needs.
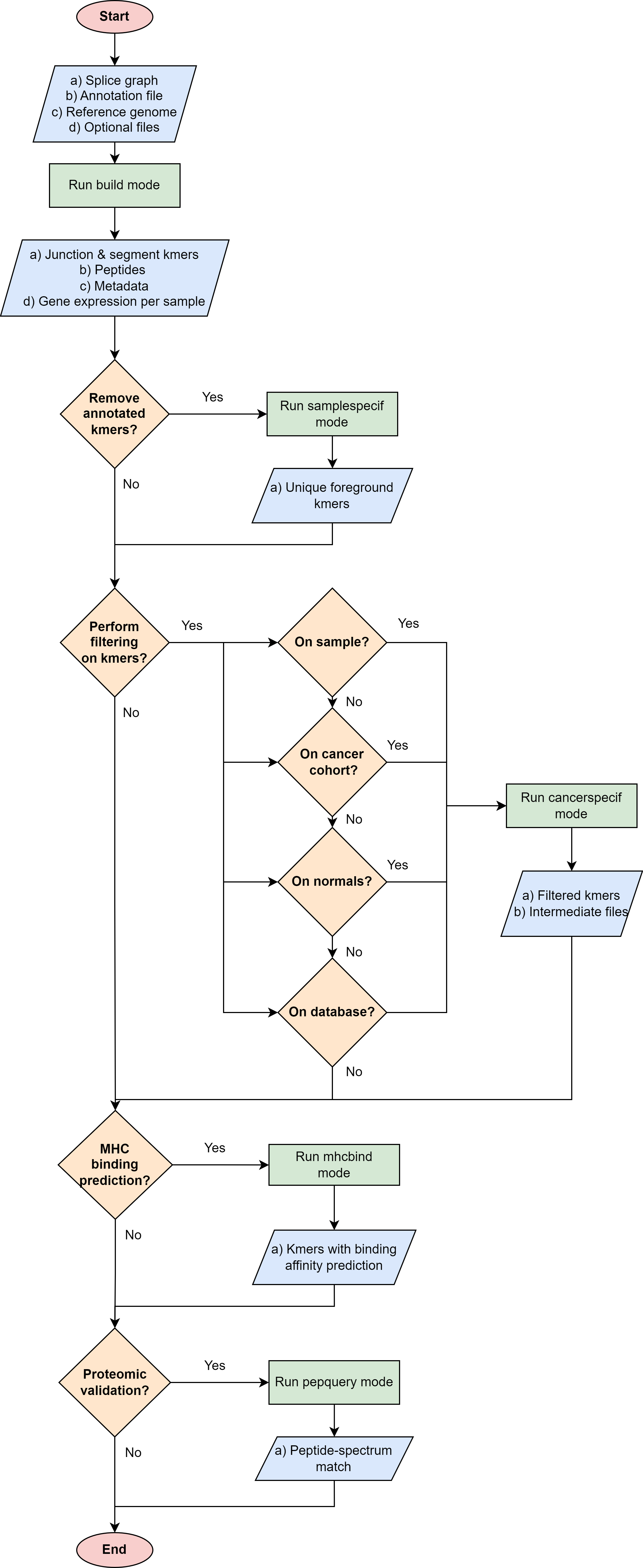
Build mode
The mode build is the core part of ImmunoPepper. It traverses the input splice graph and generates all possible peptides/kmers.
Throughout the description of the different parameters we will use two terms worth describing:
Background: This refers to the baseline set of full transcripts found in the organism as described by the annotation file provided under –ann-path. In this mode, the complete sequence of exons for each given transcript will be obtained from the annotation file. The regions corresponding to this exons will be taken from the reference genome file provided under –ref-path, and they will be translated to create the set of background peptides or kmers. In the output, background files are referred to as annot. If the user chooses to provide germline variants under –germline, the nucleotide variations will be applied to the sequence, and they will therefore be reflected in the set of background peptides and kmers.
Foreground: It aims at representing the novelty found in the organism. The software focuses on short-range novelty, as it only extracts pairs of two exons. In the case where the two exons are not enough to create a kmer of length –kmer, the software will use an exon triplet (unless –disable-concat is set to True). This mode will extract the exon pairs, or triplets, belonging to each transcript by traversing the splicing graph provided under –splice-path. Then, it will extract the sequence corresponding to the exon coordinates from –ref-path, and it will be translated to generate foreground peptides or kmers. If the user chooses to provide germline variants under –germline or somatic mutations under –somatic, the nucleotide variations will be applied to the sequence, and they will therefore be reflected in the set of foreground peptides and kmers. In the output, foreground files are referred to as sample. Moreover, the sample names will contain a prefix indicating the mutation mode that was applied, namely ‘ref’ (if no mutations were applied), “germline”, “somatic” or “somatic_and_germline”.
Note
The current implementation of SplAdder uses the provided annotation as a backbone splice graph and then adds the alternative splicing events found in the RNA-Seq data. Therefore, not all the peptides labeled as foreground will be novel, as some can be obtained from baseline exon pairs/triplets.
usage: immunopepper build [-h] --output-dir OUTPUT_DIR --ann-path ANN_PATH --splice-path SPLICE_PATH --ref-path REF_PATH --kmer KMER [--libsize-extract] [--all-read-frames] [--count-path COUNT_PATH] [--output-samples OUTPUT_SAMPLES [OUTPUT_SAMPLES ...]] [--heter-code {0,2}] [--compressed] [--parallel PARALLEL] [--batch-size BATCH_SIZE] [--pickle-samples PICKLE_SAMPLES [PICKLE_SAMPLES ...]] [--process-chr PROCESS_CHR [PROCESS_CHR ...]] [--complexity-cap COMPLEXITY_CAP] [--genes-interest GENES_INTEREST] [--start-id START_ID] [--process-num N] [--skip-annotation] [--keep-tmpfiles] [--output-fasta] [--force-ref-peptides] [--filter-redundant] [--kmer-database KMER_DATABASE] [--gtex-junction-path GTEX_JUNCTION_PATH] [--disable-concat] [--disable-process-libsize] [--mutation-sample MUTATION_SAMPLE] [--germline GERMLINE] [--somatic SOMATIC] [--sample-name-map SAMPLE_NAME_MAP] [--use-mut-pickle] [--verbose VERBOSE]Mandatory arguments
- --output-dir
absolute path of the output directory. All output files generated by immunopepper will be saved in this directory.
Default: “output”
- --ann-path
absolute path for the annotation file. Accepted file formats: .gtf, .gff and .gff3. Annotation files can be downloaded from various databases such as GENCODE
- --splice-path
absolute path of the input SplAdder splice graph.
- --ref-path
absolute path of the reference genome file in FASTA format. Reference Please ensure that the reference genome is compatible with the gene annotation file being used. For example, if the annotation file is based on GRCh38, the reference genome should also be based on GRCh38. You can check here gencode annotation releases and their corresponding major genome assembly releases. For example, if you decide to use genome assembly version GRCh38.p13, you need to use its compatible annotation file from release 43 in GENCODE.
- --kmer
length of the kmers for kmer output.
Default: 9
Submodes parameters
Commands for conceptual information about the processing.
- --libsize-extract
Set this parameter to True to generate library sizes and gene quantifications and skip neontigen generation. Note: If set to True, the program will only output files 3 and 7 of the build output section.
Default: False
- --all-read-frames
Set this parameter to True to switch to exhaustive translation and study all possible reading frames instead of just the annotated ones in the annotation file.
Default: False
- --count-path
Absolute path for the second output of SplAdder containing the graph expression quantification. If provided, expression quantification of genes will take place. Format: hdf5.
- --output-samples
List of sample names to output. Note: Names should match the file name of the splice graphs. If not provided all samples are processed and program runs faster.
Default: []
- --heter-code
Possible choices: 0, 2
It specifies the heterozygous allele.
Default: 0
Technical parameters
Commands for optimization of the software.
- --compressed
Compress output files
Default: True
- --parallel
Number of cores to be used.
Default: 1
- --batch-size
Number of genes to be processed in each batch. If bigger batches are selected, the program will be faster, but it will require more memory.
Default: 1000
- --pickle-samples
List of samples to be pickled. Needed if –use-mut-pickle is set to True. It will create intermediate files containing mutation information of the selected samples. This command is useful because mutation/variant information needs to be parsed in every run of the software, which is a time-consuming operation. By pickling the mutations, when mutation information is needed, it will be directly loaded from the intermediate pickled files instead than from the original mutation files provided under –somatic and –germline. This will speed up software re-runs. When dealing with large cohorts, this command is useful to select exactly what files should be pickled. If not provided, all the samples passed in –mutation-sample will be pickled. Names should match the sample names of the graph/counts files but an equivalence can be set using –sample-name-map from mutation parameters.
Default: []
- --disable-concat
Disable the generation of kmers from combinations of more than 2 exons. In this mode, any kmer shorter than –kmer will be discarded. By setting this command to False, the generation of kmers from 3 exons is allowed. This might ensure that kmers generated from shorter exons are kept, but one should take into account that kmers translated from 3 exons might have lower confidence. By setting the argument to True the generation of kmers is faster.
Default: False
- --disable-process-libsize
Set to True to generate the libsize file (file 7 from the build output section).
Default: False
Subset parameters
Commands to select a subset of the genes to be processed.
- --process-chr
List of chromosomes to be processed. If not provided all chromosomes are processed. The chromosomes names should be provided in the same format as in FASTA and annotation files. For annotations downloaded from GENCODE, this format is chrX, X being the chromosome number.
- --complexity-cap
Maximum edge complexity of the graph to be processed. If not provided all graphs are processed.
- --genes-interest
Genes to be processed. Format: Input is a csv file containing a gencode gene id per line, with no header. Technical note: The gencode gene id must match the gencode gene ids of the splice graph. Therefore, the format for this argument must match the format of the gene_id field of the annotation file used in the build mode of SplAdder and passed under –ann-path in immunopepper. If not provided all genes are processed.
- --start-id
Id of the first gene in the splice graph to be processed. If not provided all genes are processed.
Default: 0
- --process-num
Number of genes to be processed. If provided, the first process-num genes are processed.
Default: 0
Output parameters
Commands to select output formatting and filtering.
- --skip-annotation
Set this parameter to True to skip the generation of a background kmer and peptide files.
Default: False
- --keep-tmpfiles
If set to True, we will keep the intermediate directories and temporal files generated in paralell mode
Default: False
- --output-fasta
Set this parameter to True to output the foreground peptides in fasta format. If set to True output number 4 from the build output section will be generated. If set to False only the kmers will be output.
Default: False
- --force-ref-peptides
Set this parameter to True to output mutated peptides even if they are the same as the ones in the reference. The reference in this case are the peptides without any mutations or variants application.
Default: False
- --filter-redundant
If set to true, a redundancy filter will be applied to the exon list. If two or more exons span the same juction, their coordinates will be combined so that the longest spanning combination is kept.
Default: False
- --kmer-database
Absolute path of a file containing kmers in one column, without header. If the kmers contained in this database contain the aminoacid isoleucine (I), it will be converted into leucine (L). A file from uniprot or any other standard library can be provided. The kmers provided in this file will not be output if found in the foreground peptides. Please note that is a standard proteome is downloaded from an online resource the proteins should be cut into the kmers length selected under –kmer.
- --gtex-junction-path
- Absolute path of whitelist junction path. The junctions of this file will be the only ones output from the tool.
Format: hdf5 file with ‘chrm’, ‘pos’ and ‘strand’ as keys. ‘Chrm’ contains the chromosome name in the same format as in the annotation. ‘pos’ contains coordinates. The coordinates are end_e1 and start_e2. This means that end_e1 > start_e2 if strand is ‘-’ and end_e1 < start_e2 if strand ‘+’. strand will be either ‘-’ or ‘+’.
Pipeline relevance: When a whitelist file is provided, the field ‘isJunctionList’ in the metadata output file will contain a 1 if the junction is contained in this list, and 0 otherwise.
Mutation parameters
Commands to add somatic and germline mutation information.
- --mutation-sample
Sample id of the files to which mutations are added. The ids should match the graphs/counts names, but an equivalence can be set with –sample-name-map.
- --germline
Absolute path of the germline mutation file. Format: VCF, MAF or h5.
Default: “”
- --somatic
Absolute path of the somatic mutation file. Format: VCF, MAF or h5.
Default: “”
- --sample-name-map
Name mapping to sample names from graphs/counts files. Format: No header. Two columns: [name of count/graphs file name of mutation/pickle file]. Three columns: [name of count/graphs file name of germline file name of somatic file].
- --use-mut-pickle
Set to True to save and use pickled mutation dictionary. This command is useful because mutation/variant information needs to be parsed in every run of the software, which is a time-consuming operation. By pickling the mutations, when mutation information is needed, it will be directly loaded from the intermediate pickled files instead than from the original mutation files provided under –somatic and –germline. This will speed up software re-runs.
Default: False
GENERAL
- --verbose
specify output verbosity (0 - warn, 1 - info, 2 - debug) [1]
Default: 1
Samplespecif mode
usage: immunopepper samplespecif [-h] --annot-kmer-files ANNOT_KMER_FILES [ANNOT_KMER_FILES ...] --output-dir OUTPUT_DIR --junction-kmer-files JUNCTION_KMER_FILES [JUNCTION_KMER_FILES ...] --bg-file-path BG_FILE_PATH --output-suffix OUTPUT_SUFFIX [--remove-bg] [--verbose VERBOSE]Mandatory arguments
- --annot-kmer-files
List of absolute paths to the annotation kmer files. The files should have the name [mut_mode]_annot_kmer.gz (Files 2 from the build output section)
Default: “”
- --output-dir
Path to the output directory.
- --junction-kmer-files
Absolute path to the folder containing the foreground kmers. The possible folders one can use for this mode are: [mut_mode]_graph_kmer_JuncExpr or [mut_mode]_graph_kmer_SegmExpr (Files 5 and 6 from the build output section)
Default: “”
- --bg-file-path
Absolute path to the intermediate pooled annotation file. This file is the set of unique kmers in –annot-kmer-files files. If the file is not provided it will be generated. In order to be generated one needs to provide the folder and the file name where the file will be saved. Note: It should be a non existent folder. Format: One column with header ‘kmer’.
Default: “”
- --output-suffix
Suffix to be appended to the filtered –junction-kmer-files, e.g. samplespecif
Default: “no-annot”
Optional argument
- --remove-bg
Set to True to remove from –junction-kmer-files the kmers in –bg-file-path . If set to False, a new column is_neo_flag will be added to the –junction-kmer-files files. The column will contain False if the kmer is in –bg-file-path and True otherwise.
Default: False
GENERAL
- --verbose
specify output verbosity (0 - warn, 1 - info, 2 - debug) [1]
Default: 1
Cancerspecif mode
Note
This mode uses JAVA. In order to run it, the user needs to have JAVA installed in the system. This can be checked by running:
java -versionThis mode performs different filtering steps to keep only the kmers that are specific to a cancer sample or a cancer cohort. The user can provide different cancer and normal samples to this filtering step.
Cancer samples: These are the files that contain the kmers from the cancer sample. These files correspond to the output 5 and output 6 of the build mode output section. The user can choose whether to do the filtering in the kmers derived from segments (output 5) or in the kmers derived from junctions (output 6).
Normal samples: These are the files that contain the kmers from the control sample, i.e. the normal tissue. These files correspond to the output 5 and output 6 of the build mode output section. The user can choose whether to do the filtering in the kmers derived from segments (output 5) or in the kmers derived from junctions (output 6).
The steps for the filtering pipeline implemented in this mode will be explained in the following sections. Note: The operations performed for normal and cancer samples explained separately, reason why some parameters appear several times.
Pipeline for filtering normal samples (Optional):
Preprocessing steps: Before the filtering steps, the kmers from the normal samples are preprocessed throughout different steps.
(Obligatory) NaNs removal: The entries containing NaNs in the expression matrix are set to zero
(Obligatory) Remove the kmers appearing only in the annotation file but not in the samples: The kmers that are present in the annotation file (either junctionAnnotated or ReadFrameAnnotated are True) but have expression equal to zero across all samples are removed.
(Optional) Filter for neo-junctions: If the argument –filterNeojuncCoord is set, only the kmers belonging to novel junctions are selected. This means that only the kmers with junctionAnnotated = False will be selected. The parameter takes different input vales indicating in which dataset this filter will be applied. Filtering only on the normal cohort can be obtained by setting the parameter to ‘N’, while filtering on both normal and cancer datasets can be obtained by setting the parameter to ‘A’ (‘A’: all). Note: This is an advanced parameter. It might change in future versions of the software.
(Optional) Filter for annotated reading frames: If the argument –filterAnnotatedRF is set, only the kmers with a reading frame present in the annotation file are selected. This means that only the kmers with ReadFrameAnnotated = True will be selected, discarding the kmers that were obtained by propagating the reading frame along the splice graph. The parameter takes different input vales indicating in which dataset this filter will be applied. Filtering only on the normal cohort can be obtained by setting the parameter to ‘N’, while filtering on both normal and cancer datasets can be obtained by setting the parameter to ‘A’ (‘A’: all). Note: This is an advanced parameter. It might change in future versions of the software.
(Optional) Filter for whitelist samples: If –whitelist-normal is provided, only the selected samples will be retrieved and further studied.
Filtering steps:
Pipeline relevance: This mode aims at removing a set of normal kmers from a set of cancer kmers. The software supports flexibility in the definition of the normal cohort to remove. Normal kmers “sufficiently expressed” or “sufficiently recurrent” are included in the normal cohort and will be subsequently removed. All kmers below these thresholds will not be filtered out from the cancer kmers.
Technical use: The inclusion of normal kmers is based on two different criteria. The first one aims at setting the minimum expression in any normal sample (a), and the second aims at setting the minimum recurrence at any read level in the normal cohort (b). The two criteria below can be set independently. The user can apply (a), (b), (a) and (b), or choose not to filter on a normal cohort.
Details on the filtering steps:
(Optional) Filter for expression: If a normal kmer has expression above or equal to the threshold –cohort-expr-support-normal, the kmer is selected. As it has expression higher than the given threshold in at least one sample from the normal cohort, it cannot be considered as a cancer-specific kmer. Therefore, it is saved as a “sufficiently expressed” normal kmer, and it will be removed from the cancer samples.
(Optional) Filter for number of samples: If a kmer is expressed with any read level, i.e. Expression >0, in more than –n-samples-lim-normal samples, it is selected. As it is found in a number of samples higher than the given threshold, it cannot be considered as a cancer-specific kmer. Therefore, it is saved as a “sufficiently recurrent” normal kmer, and it will be removed from the cancer samples.
Combination of the two filtering steps into a single normal database: The kmers that are selected in the two filtering steps (a) and (b) are combined into a single database. This database will be used to filter the cancer samples.
(Optional) Filtering with external resources:
Pipeline relevance: In addition to the “threshold filtering” described above, the user may input a database of normal kmers which will be strictly subtracted from the cancer kmers. These normal kmers can be provided with the argument –path-normal-kmer-list.
Technical use:
–path-normal-kmer-list can be provided in addition to the normal database (3) obtained from the two “threshold filtering” steps described above. Both will be removed from the cancer set.
The parameter can also be provided alone. In this case, only a strict filtering of normal kmers against cancer kmers will be performed, without any “threshold filtering”.
If the parameter is not provided, the other filtering steps requested by the user will be performed.
Pipeline for filtering cancer samples:
Preprocessing steps: Before the filtering steps, the kmers from the cancer samples are preprocessed throughout different steps.
(Obligatory) NaNs removal: The entries containing NaNs in the expression matrix are set to zero
(Optional) Filter for neo-junctions: If the argument –filterNeojuncCoord is set, only the kmers belonging to novel junctions are selected. This means that only the kmers with junctionAnnotated = False will be selected. The parameter takes different input vales indicating in which dataset this filter will be applied. Filtering only on the cancer cohort can be obtained by setting the parameter to ‘C’, while filtering on both normal and cancer datasets can be obtained by setting the parameter to ‘A’ (‘A’: all). Note: This is an advanced parameter. It might change in future versions of the software.
(Optional) Filter for annotated reading frames: If the argument –filterAnnotatedRF is set, only the kmers with a reading frame present in the annotation file are selected. This means that only the kmers with ReadFrameAnnotated = True will be selected, discarding the kmers that were obtained by propagating the reading frame along the splice graph. The parameter takes different input vales indicating in which dataset this filter will be applied. Filtering only on the cancer cohort can be obtained by setting the parameter to ‘C’, while filtering on both normal and cancer datasets can be obtained by setting the parameter to ‘A’ (‘A’: all). Note: This is an advanced parameter. It might change in future versions of the software.
(Optional) Filter for whitelist samples: If –whitelist-cancer is provided, only the selected samples will be retrieved and further studied.
Filtering steps:
Pipeline relevance: This mode aims at removing a set of normal kmers from a set of cancer kmers. The software enables the user to decide at which confidence level the cancer kmers should be included. Cancer kmers can be requested to pass a user-defined expression level in one cancer sample of interest (a). Besides, if additional cancer samples are available, the user can request a kmer to be recurrent with a certain number of reads in the other cohort samples (b). All kmers below these thresholds will be filtered out.
Technical use: The user can request the “expression in a sample of interest” (a) and “cancer cohort filtering” (b) level independently, or just apply one filtering criteria. If “cancer cohort filtering” is performed (step b), expression needs to be provided in the form of a matrix.
Details on the filtering steps:
(Obligatory) Sample specific filtering: Following the preprocessing, sample-specific filtering is performed. Each sample of interest is filtered according to an expression threshold set by –sample-expr-support-cancer. The IDs of the samples of interest need to be provided with –ids-cancer-samples. For each individual sample, only the kmers with an expression level >= –sample-expr-support-cancer are selected. If –sample-expr-support-cancer is set to 0, only the kmers > –sample-expr-support-cancer are selected.
(Obligatory) Cohort filtering: After the sample specific filtering, if the cancer files are part of a cohort of patients one can do cross sample filtering. This means that the kmers that are present in more than n samples, n being the value of –n-samples-lim-cancer, with an expression higher or equal than –sample-expr-support-cancer will be selected. If –n-samples-lim-cancer is set to 0, only the kmers with an expression level > –cohort-expr-support-cancer will be selected.
Combination of the two filtering steps into a single cancer database. Kmers will be selected as cancer specific kmers if they pass both filtering steps (a) and (b), i.e. an intersection of the two filtering steps. By setting –cancer-support-union, one can select the kmers that passed either one of them or both of them, i.e. a union of the two filtering steps.
Differential filtering: The kmers appearing in the normal database will be removed from the cancer kmers. This step is performed to remove the kmers that are not specific to the cancer samples.
Filtering with external resources: If –uniprot is provided, the kmers in the file will be removed from the cancer database. If –uniprot is not provided, the cancer database will be used as it is.
usage: immunopepper cancerspecif [-h] --cores CORES --mem-per-core MEM_PER_CORE --parallelism PARALLELISM --kmer KMER --output-dir OUTPUT_DIR [--out-partitions OUT_PARTITIONS] [--scratch-dir SCRATCH_DIR] [--interm-dir-norm INTERM_DIR_NORM] [--interm-dir-canc INTERM_DIR_CANC] [--ids-cancer-samples IDS_CANCER_SAMPLES [IDS_CANCER_SAMPLES ...]] [--mut-cancer-samples {ref,somatic,germline,somatic_and_germline} [{ref,somatic,germline,somatic_and_germline} ...]] [--whitelist-normal WHITELIST_NORMAL] [--whitelist-cancer WHITELIST_CANCER] [--path-cancer-libsize PATH_CANCER_LIBSIZE] [--path-normal-libsize PATH_NORMAL_LIBSIZE] [--normalizer-cancer-libsize NORMALIZER_CANCER_LIBSIZE] [--normalizer-normal-libsize NORMALIZER_NORMAL_LIBSIZE] [--output-count OUTPUT_COUNT] [--tag-normals TAG_NORMALS] [--tag-prefix TAG_PREFIX] [--path-normal-matrix-segm PATH_NORMAL_MATRIX_SEGM [PATH_NORMAL_MATRIX_SEGM ...]] [--path-normal-matrix-edge PATH_NORMAL_MATRIX_EDGE [PATH_NORMAL_MATRIX_EDGE ...]] [--n-samples-lim-normal N_SAMPLES_LIM_NORMAL] [--cohort-expr-support-normal COHORT_EXPR_SUPPORT_NORMAL] [--sample-expr-support-cancer SAMPLE_EXPR_SUPPORT_CANCER] [--cohort-expr-support-cancer COHORT_EXPR_SUPPORT_CANCER] [--n-samples-lim-cancer N_SAMPLES_LIM_CANCER] [--path-cancer-matrix-segm PATH_CANCER_MATRIX_SEGM [PATH_CANCER_MATRIX_SEGM ...]] [--path-cancer-matrix-edge PATH_CANCER_MATRIX_EDGE [PATH_CANCER_MATRIX_EDGE ...]] [--cancer-support-union] [--path-normal-kmer-list PATH_NORMAL_KMER_LIST [PATH_NORMAL_KMER_LIST ...]] [--uniprot UNIPROT] [--filterNeojuncCoord {C,N,A}] [--filterAnnotatedRF {C,N,A}] [--tot-batches TOT_BATCHES] [--batch-id BATCH_ID] [--on-the-fly] [--verbose VERBOSE]Mandatory arguments
These arguments belong to three main groups:
Technical parameters: Due to the heavy amount of data, this mode uses spark. These are parameters to control spark processing
Input helper parameters: These parameters are used for parsing the input files by the software
General output files: Parameters for the files that are output by the software regardless of the filtering method.
- --cores
Technical parameter. Number of cores to use
Default: “”
- --mem-per-core
Technical parameter. Memory required per core
- --parallelism
Technical parameter. Parallelism parameter for Spark Java Virtual Machine (JVM). It is the default number of partitions in RDDs returned by certain transformations. Check –spark.default.parallelism here for more information.
Default: 3
- --kmer
Input helper parameter. Kmer length
- --output-dir
General output file. Absolute path to the output directory to save the filtered data.
Default: “”
Optional technical parameters
Due to the heavy amount of data, this mode uses spark. These are parameters to control spark processing
- --out-partitions
This argument is used to select the number of partitions in which the final output file will be saved. If not provided, the results are saved in a single file
- --scratch-dir
Os environment variable name containing the cluster scratch directory path. If specified, all the intermediate files will be saved to this directory. If not specified, the intermediate files will be saved to the output directory, specified under –output-dir. If the scratch directory is provided, –interm-dir-norm and –interm-dir-cancer are ignored.
Default: “”
- --interm-dir-norm
Custom scratch directory path to save the intermediate files for the normal samples. If not specified, the intermediate files will be saved to the output directory, specified under –output-dir.
Default: “”
- --interm-dir-canc
Custom scratch directory path to save the intermediate files for the cancer samples. If not specified, the intermediate files will be saved to the output directory, specified under –output-dir.
Default: “”
Optional input helper parameters
These parameters are used for parsing the input files by the software.
- --ids-cancer-samples
List with the cancer sample IDs of interest. The samples should be contained in the input cancer expression matrix, and follow the same format.
Default: “”
- --mut-cancer-samples
Possible choices: ref, somatic, germline, somatic_and_germline
List of mutation modes corresponding to each cancer sample. They will be used to tag the output files. The list should have the same number of entries as –ids-cancer-samples.
Default: “”
Optional general input files
Parameters for the input files that are used regardless of the filtering method.
- --whitelist-normal
File containing the whitelist of normal samples. If provided, only the samples in the whitelist will be retrieved and further studied. Format: Tab separated file without a header, with a single column containing sample names.
- --whitelist-cancer
File containing the whitelist of cancer samples. If provided, only the samples in the whitelist will be retrieved and further studied. Format: Tab separated file without a header, with a single column containing sample names.
- --path-cancer-libsize
Path for the libsize file of the selected cancer samples. It corresponds to the output 7 of build output section
- --path-normal-libsize
Path for the libsize of the selected normal samples. It corresponds to the output 7 of build output section
- --normalizer-cancer-libsize
Default = median of the libsize across all input samples. Custom normalization factor for the cancer libsize. Normalization is used to make all the samples comparable and correct for possible biases in data acquisition. The normalization is done according to the following formula: (read count / 75th quantile in sample) * normalizer value.
- --normalizer-normal-libsize
Default = median of the libsize across all input samples. Custom normalization factor for the normal libsize. Normalization is used to make all the samples comparable and correct for possible biases in data acquisition. The normalization is done according to the following formula: (read count / 75th quantile in sample) * normalizer value.
Optional general output files
Parameters for the files that are output by the software regardless of the filtering method.
- --output-count
Path and name where the intermediate numbers of kmers remaining after each filtering step will be written. If selected, a file will be written containing the number of kmers present after each filtering step. It might slow down the computations. However, it is useful if there is an interest on intermediate filtering steps. The output can be seen here in the output section.
Default: “”
- --tag-normals
Name for the normal cohort used for filtering. Needed when there are various normal cohorts. It will be added to the final output name in order to identify the normal cohort against which the cancer samples were filtered.
Default: “”
- --tag-prefix
Prefix used for the output files. It is recommended when there are different conditions being studied.
Default: “”
Optional parameters for normal samples filtering
Parameters to perform the step 2 of the normal filtering pipeline.
- --path-normal-matrix-segm
Path to the matrix of segment expression of kmers in normal samples. This corresponds to output 5 of build output section
- --path-normal-matrix-edge
Path to the matrix of junction expression of kmers in normal samples. This corresponds to output 6 of build output section
- --n-samples-lim-normal
This is the value for threshold b) in step 2 of normal filtering pipeline. This number will set the number of samples in which we need to see any expression of a specific kmer, i.e. expression > 0, to consider it a normal kmer and exclude it as cancer candidate.
- --cohort-expr-support-normal
This is the value for threshold a) in step 2 of normal filtering pipeline. This number corresponds to the normalized expression we need to see in at least one sample in order to consider a kmer as a normal kmer. If a kmer is found with an expression level higher or equal than this threshold in one or more normal samples it will be considered a normal kmer and excluded as cancer candidate.
Optional parameters for cancer samples filtering
Parameters to perform the step 2 of the cancer filtering pipeline.
- --sample-expr-support-cancer
This parameter corresponds to sample specific filtering in the cancer pipeline. The value will correspond to the normalized expression threshold that needs to be reached by each kmer in order to be considered a kmer candidate. Kmers with an expression higher or equal than this threshold in the cancer sample of interest will be considered as cancer candidates. Cancer samples of interest are provided under –ids_cancer_samples.
- --cohort-expr-support-cancer
This parameter corresponds to the expression threshold in cohort specific filtering. It indicates the normalized expression value that needs to be observed in at least –n-samples-lim-cancer in order to consider a kmer as a cancer candidate. Kmers with an expression higher than this threshold in at least –n-samples-lim-cancer samples will be considered as cancer candidates. For each cancer sample of interest, provided under –ids_cancer_samples, the expression threshold –cohort-expr-support-cancer will be assessed in the rest of the cohort, excluding the sample of interest.
- --n-samples-lim-cancer
This parameter corresponds to the number of samples threshold in cohort specific filtering. It indicated the minimum number of cancer samples in which one should see an expression higher than –cohort-expr-support-cancer in order to consider the kmer as a cancer candidate. Kmers with an expression higher than –cohort-expr-support-cancer in at least –n-samples-lim-cancer samples will be considered as cancer candidates. For each cancer sample of interest, provided under –ids_cancer_samples, the expression threshold –cohort-expr-support-cancer will be assessed in the rest of the cohort, excluding the sample of interest.
- --path-cancer-matrix-segm
Path to the cancer matrix containing segment expression from samples belonging to a cohort. The matrix will have the following dimensions: [kmers * samples]. When only the junction overlapping cancer kmers are of interest, the user should provide only –path-cancer-matrix-edge, and skip the inclusion of this file.If both matrices are provided, junction expression will be chosen in case there is expression information for the same kmer in both matrices. This will be the output 5 of build output section
- --path-cancer-matrix-edge
Path to the cancer matrix containing junction expression from samples belonging to a cohort. The matrix will have the following dimensions: [kmers * samples]. When only the junction overlapping cancer kmers are of interest, the user should provide only this file, and skip the inclusion of –path-cancer-matrix-segm. If both matrices are provided, junction expression will be chosen in case there is expression information for the same kmer in both matrices. This will be the ouput 6 of build output section.
- --cancer-support-union
Parameter to choose how the sample specific filtering and the cohort specific filtering are combined. By default, they are combined by choosing the common kmers to both filtering steps, i.e. performing an intersection. If this parameter is set to True, the union of both filtering steps will be performed, i.e. the kmers that pass either the sample specific filtering or the cohort specific filtering will be kept.
Default: False
Optional parameters for the addition of additional backgrounds
Parameters to add additional backgrounds that will be removed.
- --path-normal-kmer-list
List of kmers to be added as part of the normal samples. The kmers provided in this list will be included in the normal background, without having to pass any of the filtering steps for the normal data. Format: It can be either a tsv or parquet file. If parquet is the used format, the file should contain the header ‘kmer’ in the first column.
- --uniprot
Path to file containing uniprot kmers. The kmers contained in this databased will be assumed to be not novel peptides, and they will be removed from the cancer filtering output. Note: It is important to kmerize the peptides downloaded from uniprot database into the length specified in `–kmer`.
Optional parameters to add additional filters
Parameters to add the additional filters shown in step 1 of both pipelines.
- --filterNeojuncCoord
Possible choices: C, N, A
This argument will filter the neojunctions, and it will retain only the kmers that are generated from neojunctions. These are peptides whose junction coordinates were not part of the annotation files. Selecting this option means that only those kmers with junctionAnnotated = False will be considered for further filtering. This filter is used in the preprocessing (step 1 of cancer and normal pipelines). If ‘C’ is selected, the filter is only applied to the cancer sample. If ‘N’ is selected, the filter is only applied to the normal samples. If ‘A’ is selected, the filter is applied to both cancer and normal samples.
Default: “”
- --filterAnnotatedRF
Possible choices: C, N, A
This argument will only retrieve kmers that were generated from annotated reading frames and discard those that were obtained by reading frame propagation through the graph. By selecting this option, only kmers generated from reading frames present in the annotation are kept. This means that selecting this option means that only those kmers with ReadFrameAnnotated = True will be selected. This filter is used in the preprocessing (step 1 of cancer and normal pipelines). If ‘C’ is selected, the filter is only applied to the cancer sample. If ‘N’ is selected, the filter is only applied to the normal samples. If ‘A’ is selected, the filter is applied to both cancer and normal samples.
Default: “”
Optional development parameters
- --tot-batches
If selected, the filtering of the background and foreground will be based on hash functions. This parameter will set the total number of batches in which we will divide the foreground and background files to filter, and each of those batches will be assigned a hash value. If –batch-id is specified, –tot-batches should also be specified.
- --batch-id
If selected, the filtering of the background and foreground will be based on hash functions. This parameter will set the batch id of the current batch that is being filtered. The batch id should be an integer between 0 and –tot-batches. It shows the specific batch that we want to process, out of the –tot-batches. If –batch-id is specified, –tot-batches should also be specified.
- --on-the-fly
If set to true, all the filtering steps will be done on the fly, without the creation of intermediate files. The creation of intermediate files would speed up the computations in the case where full or partial reruns are planned. An example of a partial rerun would be when seveal normal and cancer filtering parameters are applied to the same cohort. Choosing not to save intermediate files will trade speed for disk space.
Default: False
GENERAL
- --verbose
specify output verbosity (0 - warn, 1 - info, 2 - debug) [1]
Default: 1
Mhcbind mode
usage: immunopepper mhcbind [-h] --mhc-software-path MHC_SOFTWARE_PATH --argstring ARGSTRING --output-dir OUTPUT_DIR [--partitioned-tsv PARTITIONED_TSV] [--bind-score-method BIND_SCORE_METHOD] [--bind-score-threshold BIND_SCORE_THRESHOLD] [--less-than] [--verbose VERBOSE]Mandatory arguments
- --mhc-software-path
Path for the MHC prediction software.
- --argstring
- Complete command line for the MHC prediction tool passed as a string. One should include here the command that will be directly passed to the selected MHC tool. The four mandatory arguments are:
1.–mhc-predictor: This argument will specify the name of the software tool that will be used. The name should be in the format accepted by the library mhc_tools
2.–output-csv: This argument will contain the path and filename where the MHC prediction tool will save the results. The format of the file should be .csv.
3.–input-peptides-file: This argument will have the path and filename to the file containing the set of kmers on which MHC binding affinity prediction will be performed. The format of the file should be .csv.
–mhc-alleles or –mhc-alleles-file: This argument should contain the alleles that will be used for the analysis of mhc binding affinity.
If –partitioned-tsv files are provided, an intermediate file will be created and stored under the path –input-peptides-file . This intermediate file will contain the set of all unique kmers present in the partitioned files obtained from cancerspecif mode. If one does not want to use the output of cancerspecif mode for prediction, the path to the file that will be used for prediction will be directly provided under –input-peptides-list.
Default: “”
- --output-dir
General output file. Absolute path to the output directory to save the MHC predictions. It should match the directory provided in –output-csv of the argstring, but without the file name.
Default: “”
Optional argument
- --partitioned-tsv
The input to this command is the path to the folder containing the partitioned tsv files from cancerspecif mode. This corresponds to the files 1 and 2 found in the output section. If this parameter is set the tool will directly accept the files from cancerspecif mode as input.
- --bind-score-method
Scoring method to filter the MHC tools predictions. E.g. score, affinity, percentile_rank (this last one is only for netmhcpan).
- --bind-score-threshold
Threshold to filter the MHC tools predictions. All the peptides with a score lower than the threshold will be filtered out and only the ones with a score higher than the threshold will be kept.
- --less-than
If set to True the –bind-score-threshold will be considered as an upper bound instead of a lower bound. This means that peptides with a score higher than this threshold will be filtered out.
Default: False
GENERAL
- --verbose
specify output verbosity (0 - warn, 1 - info, 2 - debug) [1]
Default: 1
Pepquery mode
This mode is a wrapper of the software PepQuery. It allows the user to do MS/MS based validation of the kmers identified using immunopepper.
By using this mode, the user can identify matches between the kmers of interest and a specific MS/MS spectra of interest. This is useful to perform a validation at the peptide level.
In the output of this software, several steps are referenced. The steps correspond to the filtering steps used in the PepQuery software. The different steps are:
Peptide sequence preparation and initial filtering: In this case, as the input is already a peptide or a list of peptides, there is not preparation needed. The peptide sequence input is used directly
Candidate spectra retrieval and peptide spectra match (PSM) scoring: Each peptide is searched against the provided MS/MS dataset. The candidate spectra are studied by looking at the mass difference between the peptide and each spectrum. It uses a user-specified mass tolerance, that can be set using “-tol” in the pepQuery software. The scoring is done according to one of the available metrics, which can be chosen by setting “-m” in the pepQuery software.
Competitive filtering based on reference sequences: The spectra identified in step 2 are searched against the reference database provided by the user. The spectra that have a better match with a reference peptide are removed.
Statistical evaluation: If no better match was found in the reference database, a statistical evaluation is performed. The peptides are random shuffled and the statistical significance of the match is assessed by computing the pvalue. Matches with a pvalue <0.01 are considered significant.
Competitive filtering based on unrestricted modification searching: The remaining significant matches are scored against the proteins of the reference database, but modified by several post translational modifications. If a spectra matches better a modified protein than the peptide of interest it is removed.
The PSM that pass all the filters are retrieved as confident.
Note
PepQuery is a memory intensive software. It needed 10 cpus and 78,2GB memory total and 24min for querying 7500 peptides against 25017 spectra. The user should take this into account when running the software.
Note
The user should make sure that the spectra provided contains the sample that was used to derive the input peptides.
More information can be obtained in the PepQuery paper: Wen, Bo, Xiaojing Wang, and Bing Zhang. “PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations.” Genome research 29.3 (2019): 485-493.
usage: immunopepper pepquery [-h] --output-dir OUTPUT_DIR --argstring ARGSTRING --pepquery-software-path PEPQUERY_SOFTWARE_PATH [--partitioned-tsv PARTITIONED_TSV] [--metadata-path METADATA_PATH] [--kmer-type {junctions,segments}] [--verbose VERBOSE]Mandatory arguments
- --output-dir
Absolute path to the output directory to save the results of the validation.
- --argstring
- Complete command line for the pepQuery MS based validation software passed as a string. The command provided here will be directly passed to the pepQuery software. It must contain several mandatory arguments:
-o: Output directory. The results from pepQuery will be saved in this folder.
-i: Input peptides for the validation. The user can either provide their own peptides or take the expanded peptides from the kmers provided in –partitioned-tsv. If –partitioned-tsv is provided, the expanded peptides will be saved under the path and filename provided in this argument, and they will be taken as the input of pepQuery.
-db: Path to the reference database used in the analysis. This reference database will be used to identify those MS/MS spectra that match better a reference peptide than the query peptides.
-ms: Path to the MS/MS spectra file.
- --pepquery-software-path
Path for the pepQuery software. It must include the path to the directory with the software and the software name itself. eg. pepquery/pepquery.jar
Optional argument
- --partitioned-tsv
The input to this command is the path to the folder containing the partitioned tsv files from cancerspecif mode. This corresponds to the files 1 and 2 found in the output section. If this parameter is set the tool will directly accept the files from cancerspecif mode as input. An intermediate file will be saved in the output directory containing the kmers in a bigger peptide context. This argument accepts files generated from junctions and from segments.
- --metadata-path
Absolute path to the metadata file created in build mode. This file is required whether the user chooses to work with junctions of with kmers.
- --kmer-type
Possible choices: junctions, segments
Type of the kmers introduced under –partitioned-tsv. This will show whether the user chooses to work with junctions or with segments. The peptide retrieval strategy will vary depending on the input type.
GENERAL
- --verbose
specify output verbosity (0 - warn, 1 - info, 2 - debug) [1]
Default: 1